


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
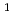 D)+N
D)+N (X
(X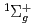 )
) O(
O( P)+N(X) spin-forbidden electronic quenching collision
P)+N(X) spin-forbidden electronic quenching collision高柳 敏幸
Journal of Physical Chemistry A, 106(19), p.4914 - 4921, 2002/05
被引用回数:19 パーセンタイル:51.36(Chemistry, Physical)O(D)+N(X)O(P)+N(X)スピン禁制衝突について量子散乱計算を行った。一重項及び三重項についてそれぞれ一枚のポテンシャル面に簡略化した。標準的な堅密結合法により、電子的非断熱消光確率及び消光断面積を計算した。ポテンシャル面及びスピン軌道相互作用については過去にほかの研究者によって作製されたものを用いた。堅密結合の計算結果は、電子的非断熱過程が量子力学的な共鳴状態を経由して起こることを示した。このことは、これまで提唱されてきたポテンシャル交差モデルが、この非断熱過程には使えないことを示している。また、堅密結合の結果を半古典的ホッピングトラジェクトリー近似法による計算結果とも比較し、この近似が不十分であることも示した。
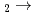 He+H+H reaction
He+H+H reaction高柳 敏幸; 和田 晃
Chemical Physics, 277(3), p.313 - 323, 2002/04
被引用回数:6 パーセンタイル:17.74(Chemistry, Physical)擬連続近似を使った3次元量子堅密結合法をHe+HHe+H+H衝突誘起解離反応に適用した。この研究の大きな目的は、反応の解離極限よりわずかに高いエネルギー領域で、共鳴を経由した2体衝突と共鳴を経由しない3体衝突のどちらが主体かを理論的に確かめることである。実際の計算は全角運動量がゼロの場合のみについて行ったが、われわれは、水素分子の回転量子数が小さい場合には、共鳴を経由しない3体衝突が支配的であり、共鳴を経由する2体衝突過程は、回転量子数が大きい場合のみに重要になることを見出した。さらに、衝突誘起解離が起こる確率は、主に水素分子の振動回転エネルギー準位と、解離極限のエネルギー差でほとんど決まることを見出した。このことは、衝突誘起解離過程は、簡単なエネルギー移動過程であることを意味するものである。並進運動を古典的に扱い、水素分子の振動回転運動を量子的に取り扱う半古典近似と今回の計算結果を比較したところ、解離確率の小さなエネルギー領域では、半古典論が全く機能しないことも確かめられた。
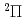 )+N(X)
)+N(X)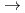 HCN(X
HCN(X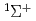 )+N(
)+N(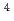 S) reaction
S) reaction和田 晃; 高柳 敏幸
Journal of Chemical Physics, 116(16), p.7065 - 7072, 2002/04
被引用回数:10 パーセンタイル:30.24(Chemistry, Physical)スピン禁制反応 CH(X)+N(X)HCN(X)+N(S) について、量子散乱理論を用いた計算を行った。CH分子を一個の原子とみなすことによって、自由度を3次元に落とした。分子軌道計算を用いて、スピン2重項及び4重項それぞれのポテンシャルエネルギー曲面を作製した。また、スピン軌道相互作用については過去の理論計算を用いた。超球座標を用いた堅密結合方程式を数値的に解いて、総反応確率を計算した。計算された確率は典型的な共鳴構造を示した。得られた確率から反応速度定数を計算し、実験結果と比較したところ、100倍ほど小さな値が得られたが、速度定数はスピン軌道相互作用に大きく依存することがわかった。
) reaction system
reaction system高柳 敏幸; 黒崎 譲; 市原 晃
Journal of Chemical Physics, 112(6), p.2615 - 2622, 2000/02
被引用回数:62 パーセンタイル:86.41(Chemistry, Physical)非断熱遷移を伴う(D+H)イオン分子反応について3次元量子散乱計算を行った。超球座標を使った時間に依存しないclose-coupling法を用いた。ポテンシャルエネルギー曲面として(3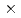 3)のDIMポテンシャルを使った。正確な量子論の計算結果を半古典的なトラジェクトリホッピングの結果と比較した。その結果Tullyによって提唱されている方法のほうが従来から使われているTully-Prestonの方法よりも量子論の結果をよく再現することがわかった。これはTully-Prestonの方法が、ポテンシャルの交差付近でのみの電子遷移しか考慮していないことが原因である。
3)のDIMポテンシャルを使った。正確な量子論の計算結果を半古典的なトラジェクトリホッピングの結果と比較した。その結果Tullyによって提唱されている方法のほうが従来から使われているTully-Prestonの方法よりも量子論の結果をよく再現することがわかった。これはTully-Prestonの方法が、ポテンシャルの交差付近でのみの電子遷移しか考慮していないことが原因である。
reaction and its isotopic variants; The effect of the Van der Waals potential高柳 敏幸; 黒崎 譲*
Chemical Physics Letters, 286(1-2), p.35 - 39, 1998/04
被引用回数:32 パーセンタイル:69.99(Chemistry, Physical)F+H,D及びHD反応について、正確なポテンシャルエネルギー曲面を用いて3次元の反応性量子散乱計算を行い、反応確率を理論的に計算した。反応確率はしきい値付近で小さなピークをもつことを見出し、これはVan der Waals井戸に形成される共鳴状態との干渉効果であることをつきとめた。F+HD反応では特に低エネルギーでHF+Dを生成する確率がトンネル効果のために支配的であることがわかった。またVan der Waals共鳴を用いて化学反応をコントロールできる可能性があることも見出した。
高柳 敏幸
Bulletin of the Chemical Society of Japan, 68(9), p.2527 - 2532, 1995/00
被引用回数:18 パーセンタイル:68.38(Chemistry, Multidisciplinary)一般的な4原子系引き抜き反応、AB+CDA+BCDについて、近似的な量子反応性散乱理論を開発した。この近似理論を用いて、具体的な反応、H+CNH+HCNの反応確率を計算した。ポテンシャルエネルギー曲面として、ab initio分子軌道計算に基づく半経験的な関数を用いた。この理論の特徴は化学反応における立体効果を定量的に見積もることができることである。得られた結果を古典的トラジェクトリー法による結果と比較したところ、定性的な一致を得た。また、反応に及ぼす振動モードの励起の影響についても検討した。
